

估计阅读时长: 2 分钟在BILIBILI上观看视频:《【GCModeller教程】基因组功能富集计算原理》 Order by Date Name Attachments 20190818_GSEA_release.mp4_20190921_225144.467 • 226 kB • 735 click 2021年5月30日Fisher Exact Test […]

估计阅读时长: 2 分钟在BILIBILI上观看视频:《【GCModeller教程】KEGG代谢途径注释原理 (重置版)》 Order by Date Name Attachments kegg_annotation • 468 kB • 829 click 2021年5月30日release.mp4_20190921_225235.396 • […]
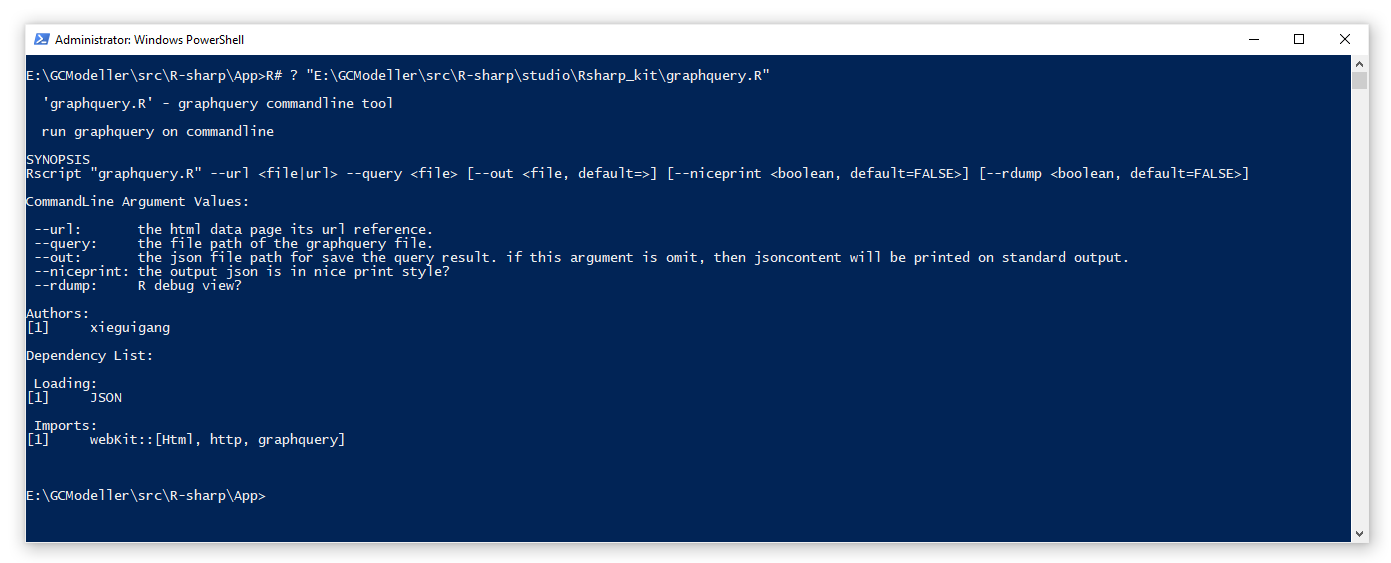
估计阅读时长: 9 分钟前段时间由于工作的需要,会需要从一些网站上抓取数据用来做数据分析。在原来我进行网页爬虫开发的时候,一般会需要专门针对网页格式,使用大量的正则表达式进行内容的解析。由于你也知道,VisualBasic语言所开发的程序为一个编译好的Assembly文件,所以假若所需要爬取的网页格式变化了,我们就需要对代码做修改和重新编译。这个时候就会非常的不方便。 Order by Date Name Attachments ea5d2885-bba5-410f-b02b-0589613412ed • 12 kB • 754 click 2021年5月29日graphquery_Rscript • 36 […]

估计阅读时长: < 1 分钟https://github.com/rsharp-lang/R-sharp R#语言最开始的开发需求来自于对GCModeller的组件的调用需求。因为最开始GCModeller使用的是命令行模式进行运行,但是因为VB.NET语言为编译型语言,所开发的应用程序在发布之后,用户无法轻易的修改。自己对于一些比较个性化的数据分析,在引入R#语言之前,需要专门编写一段命令行代码跑GCModeller,会十分的不方便。所以后面就有了R#脚本语言的开发。 R#语言类似于R或者Matlab语言,也是一种向量化的编程脚本语言。其语法源自于R语言,同时也结合了一些TypeScript的语法,例如TypeScript之中的字符串插值语法就被引入了R#语言之中。 const words = ["world", "R# language", "GCModeller User"]; const hello = `hello ${words}!`; […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?