
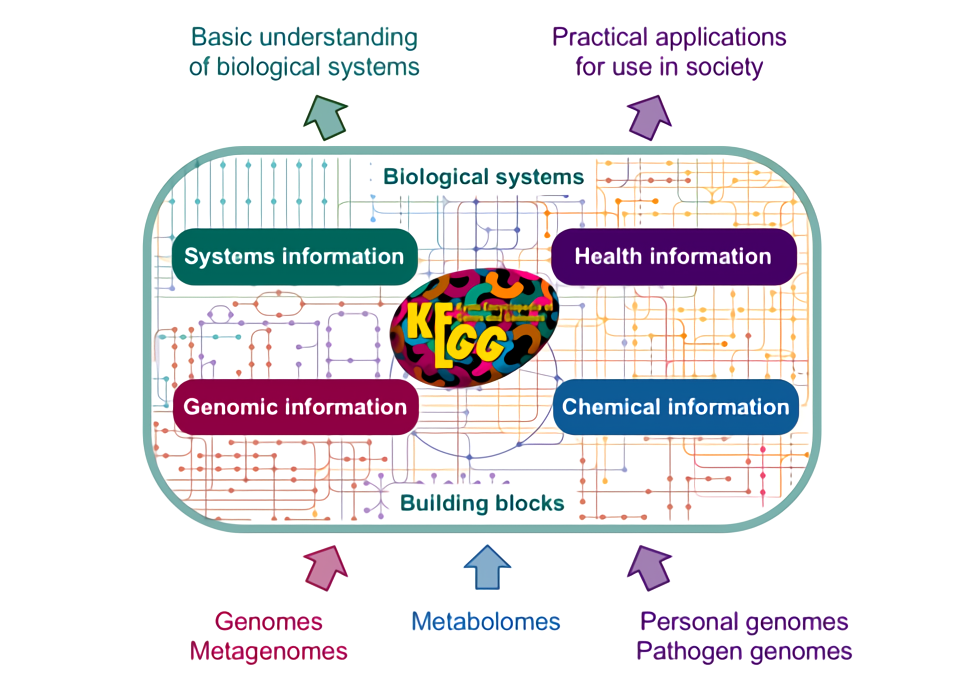
估计阅读时长: 34 分钟在前面写了一篇文章来介绍我们可以如何通过KEGG的BHR评分来注释直系同源。在KEGG数据库的同源注释算法中,BHR的核心思想是“双向最佳命中”。它比简单的单向BLAST搜索(例如,只看你的基因A在数据库里的最佳匹配是基因B)更为严格和可靠。在基因注释中,这种方法可以有效减少因基因家族扩张、结构域保守等原因导致的假阳性注释,从而更准确地识别直系同源基因,而直系同源基因通常具有相同的功能。在今天重新翻看了下KAAS的帮助文档之后,发现KAAS系统中更新了下面的Assignment score计算公式: We define a score for each ortholog group in order to assign the best […]

估计阅读时长: 27 分钟宏基因组测序直接从环境样本获取所有生物的遗传物质,产生的海量短读序列(reads)需要被快速准确地分类到不同物种或功能类别。然而,宏基因组数据具有复杂性高、物种多样且未知序列多等特点,这给分类算法带来了巨大挑战。传统的序列比对方法虽然准确,但在面对庞大的参考数据库时计算开销巨大,难以满足实时分析的需求。因此,研究者开发了多种基于k-mer(长度为k的子序列)的快速分类方法,其中布隆过滤器(Bloom Filter)作为一种高效的概率数据结构,在针对测序reads做物种上的快速分类这项工作中起到了一些关键作用。 Attachments Capture • 112 kB • 269 click 2025年12月19日
估计阅读时长: 14 分钟宏基因组测序所处理的对象是直接对环境样本中的所有DNA进行测序。达到无需培养即可揭示微生物群落的组成和功能潜力的目的。在数据处理中,一个核心任务是从海量短读序列中估算物种丰度(即每个物种在样本中的相对含量)和基因丰度(即每个基因或功能单元的相对含量)。传统的基于序列比对的方法计算成本高昂,而基于k-mer的方法通过利用固定长度的子序列(k-mer)信息,能够在不依赖完整比对的情况下快速估算丰度。 k-mer是指长度为k的连续子序列,例如在k=2的时候,DNA序列“ATCG”包含的2-mers有“AT”、“TC”、“CG”。通过统计读序列中k-mer的出现频率,并将其与参考数据库中的k-mer频率进行比较,我们可以推断出样本中各物种或基因的丰度。这种方法具有计算速度快、内存效率高的优势,并且无需对每个读进行精确比对,因此在处理大规模宏基因组数据时非常实用。 Order by Date Name Attachments workflow1 • 272 kB • 241 click 2025年12月8日workflow2 • […]
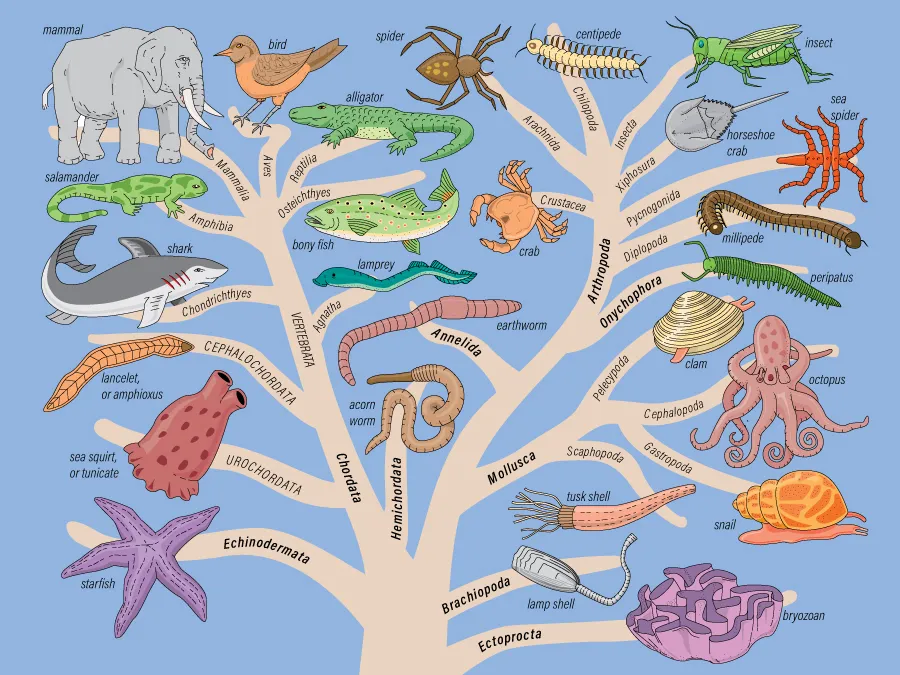
估计阅读时长: 13 分钟LCA算法是现代宏基因组学分析的核心技术之一,主要用于解决序列比对结果的分类不确定性问题。例如,我们在处理宏基因组测序reads的物种来源分类注释工作的时候,经常会思考一个问题:在宏基因组分析中,一个测序read通常与多个参考序列产生比对结果,这些结果可能指向不同的分类单元。那这条reads最可能的物种分类来源位置是怎样的,怎样可以通过一个算法,基于一系列的物种匹配结果来推断出一个合适的物种来源,既避免过度分类,又保证分类的准确性。 Order by Date Name Attachments family-tree-animal-kingdom • 99 kB • 308 click 2025年12月2日LCA • 245 […]
估计阅读时长: 2 分钟宏基因组学(Metagenomics)通过直接测序环境样本中的全部DNA,从而避免了传统培养方法的局限,使我们能够研究不可培养微生物的多样性。然而,当样本来自宿主相关环境(如人类或小鼠的肠道、土壤等)时,测序数据中不可避免地包含大量宿主自身的DNA序列。这些宿主序列会占据测序读数,增加分析成本,并可能干扰对微生物群落组成的准确推断。因此,在宏基因组数据分析中,去除宿主序列(Host Sequence Removal)是至关重要的预处理步骤。去除宿主序列的算法多种多样,其中基于k-mer的方法因其高效和可扩展性而备受关注。 Attachments Metagenomics • 211 kB • 278 click 2025年11月29日
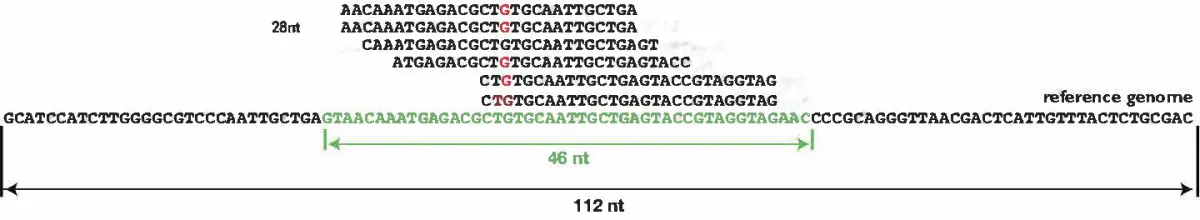
估计阅读时长: 11 分钟给定一组n个字符串数组,找到包含给定集合中每个字符串的最小字符串作为子字符串。我们可以假设这个字符串数组中没有字符串是另一个字符串的子字符串。那么基于上面的描述,我们就可以得到下面所示的问题求解目标: let arr[] = ["catg", "ctaagt", "gcta", "ttca", "atgcatc"] // output: gctaagttcatgcatc 上面的问题描述实际上是一个最短超字符串问题(shortest common superstring) Order […]

估计阅读时长: 7 分钟一般而言,进行全基因组的转录表达调控网络的建立,我们需要基于两个数据结果来完成: 目标基因的转录调控位点信息(Motif搜索结果,构成网络之中的节点) 转录调控位点相应的转录调控因子(Motif位点相关的转录调控因子,构成网络之中的边连接) Order by Date Name Attachments Xor • 271 kB • 707 click 2022年6月11日An […]
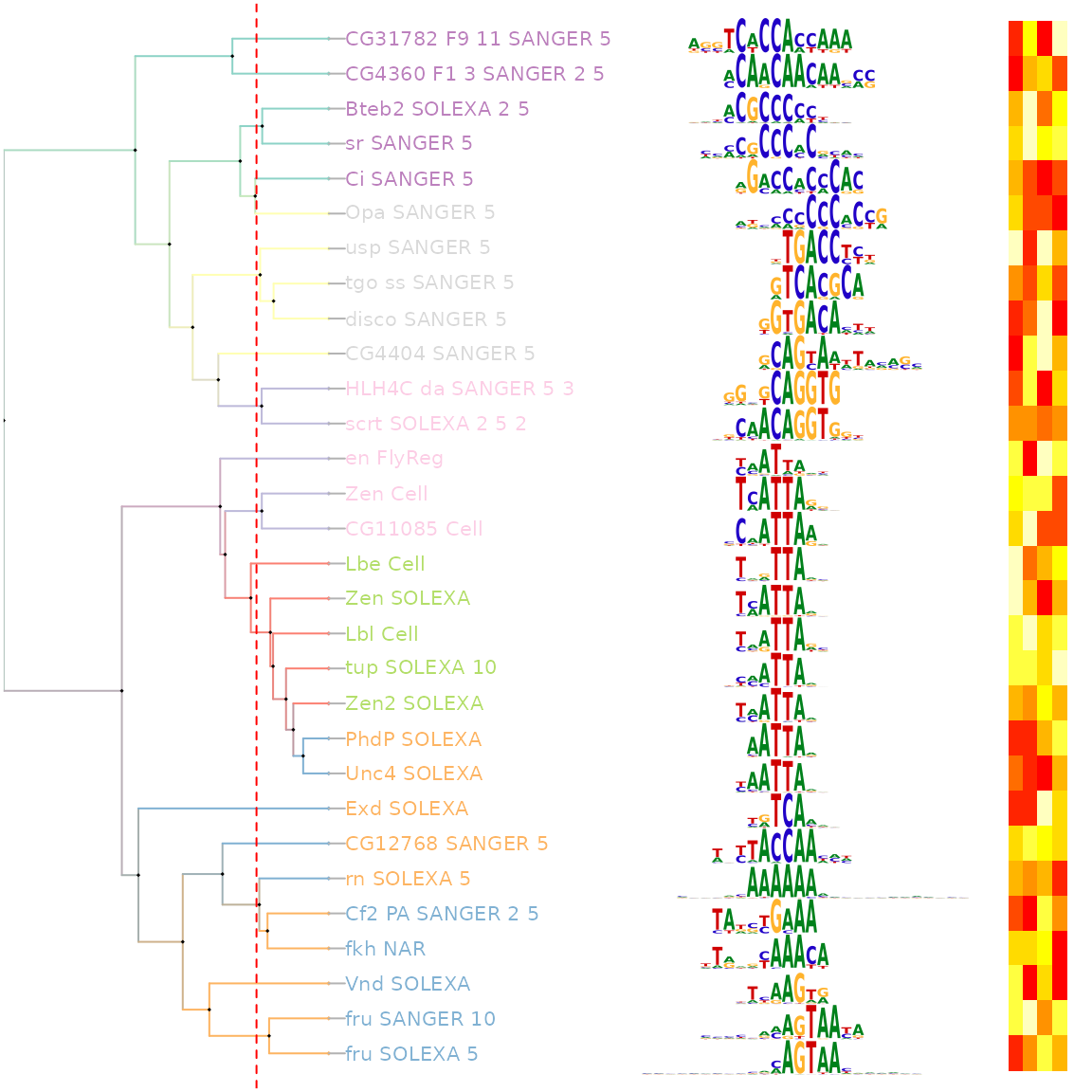
估计阅读时长: 12 分钟Motif是一段典型的序列或者一个结构。一般情况下是指构成任何一种特征序列的基本结构。通俗来讲,即是有特征的短序列,一般认为它是拥有生物学功能的保守序列,可能包含特异性的结合位点,或者是涉及某一个特定生物学过程的有共性的序列区段。比如蛋白质的序列特异性结合位点,如核酸酶和转录因子。 Order by Date Name Attachments Smith-Waterman-Algorithm-Example-Step3 • 8 kB • 706 click 2022年6月7日motifPilesHeatmap-1 • 227 […]
估计阅读时长: < 1 分钟Order by Date Name Attachments HEStainModelPreviews • 361 kB • 682 click 2022年6月3日13546516212177 • 152 kB […]
估计阅读时长: 7 分钟F统计量是群体遗传学中由Sewall Wright提出的重要统计量,用于衡量遗传变异在群体中的分布情况。它提供了对群体遗传结构和遗传分化的定量描述。F统计量主要有三种类型:Fis、Fit和Fst,分别反映个体内的、总体的和群体间的遗传分化。F统计量在群体遗传学中通常指的是Fst(Fixation Index,固定指数),它是一个衡量群体间遗传差异的指标。Fst的值范围从0到1,其中0表示群体间没有差异,1表示群体间完全分离。在群体遗传学研究中,Fst常用于评估群体的遗传多样性、群体间的迁移率以及自然选择的压力等。 Order by Date Name Attachments 41598_2021_92984_Fig1_HTML • 2 MB • 747 click 2022年5月28日p1 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?